Anaemia has been acknowledged as a frequent systemic complication and/or extraintestinal manifestation in inflammatory bowel disease (IBD)1 with iron deficiency (ID) being the primary cause of disease2 with an estimated prevalence of iron deficiency anaemia (IDA) among patients with IBD at approximately 45 %.3
Considering the potential effect on hospitalization rates and consequences for patients quality of life, anaemia has been described as a significant and costly complication of IBD.4 However, ID and IDA are under-treated.5
The PHOSPHARE-IBD study is the first randomized double-blind clinical trial to compare the incidence of hypophosphataemia after treating inflammatory bowel disease (IBD) patients diagnosed with Iron deficiency anaemia (IDA) with intravenous irons; FCM1 or FDI2,3 The results substantiate previous findings showing that patients assigned to receive FCM1 had a significantly higher incidence of hypophosphataemia compared to certain other iv iron compounds.4–6
Even when IBD patients are screened, ID and IDA might be overlooked as commonly used laboratory tests may be compromised due to several pathophysiological mechanisms related to the inflammatory nature of IBD9
Consequently, when evaluating ID and IDA in IBD the following mechanisms should be taken into consideration:
Hemoglobin: Iron deficiency should not be excluded despite normal Hb as Hb levels do not decline until a significant amount of iron is lost.10
Ferritin: Besides binding and storing iron in the liver, spleen, and reticuloendothelial system ferritin is also an acute-phase protein and can be increased in the presence of inflammation.11 Following, the interpretation of serum ferritin in patients with inflammation is tricky.5
Hepcidin and ferroportin: The hepatic peptide hormone hepcidin functions as an important regulator of iron homeostasis by controlling ferroportin – the sole iron exporter. However, hepcidin also increases as a response to inflammation.13 The increased levels of hepcidin caused by inflammation will promote the degradation of ferroportin and subsequently impair the exportation of cellular iron into plasma.13
Transferrin saturation: The main iron carrier protein is transferrin.6 In the presence of inflammation, a normal ferritin level does not exclude ID.14 In this case, testing TSAT provides an indication of the extent of iron utilization.10
Soluble transferrin saturation receptor: The TSAT level only indicates the extent of iron utilization.4 A more extensive workup should include soluble transferrin receptor (sTfR) testing.8 sTfR is a truncated form of the cellular transferrin receptor and circulates bound to transferrin.10 It is released in proportion to the expansion of erythropoiesis in the bone marrow and is not regulated by inflammation.5 As a reflector of erythropoiesis, sTfR correlates with the amount of iron available.
Hypophosphatemia is characterized by low levels of phosphate in the blood. The divergence between FCM1 and FDI2 arises due to FCM1 and FDI2’s different effects on fibroblast growth factor 23 (FGF23) affecting phosphate regulation.3 This occurs through the stimulation of renal phosphate excretion and downstream effects on 1,25-dihydroxy vitamin D and parathyroid hormone (PTH) that maintain hypophosphataemia.3
Symptoms of hypophosphataemia can be mild and are relatively non-specific, such as fatigue, which makes them challenging to differentiate from similar symptoms associated with IDA and IBD.7 However, the consequences of hypophosphatemia can extend beyond low phosphate levels with reports of complications such as severe muscle weakness and alterations in mineral and bone metabolism that can result in osteomalacia and fractures.8
The PHOSPHARE-IBD trial was conducted at 20 outpatient hospital clinics in Europe with a scope to explore the following endpoints:3
Adults with IBD and IDA (haemoglobin (Hb) <130 g/L and serum ferritin ≤100 μg/L ) were randomized 1:1 to receive either FCM1 or FDI2 treatment.3 A total of 156 patients were screened and 97 (48 FDI2, 49 FCM1) were included to receive either one dose of FCM1 or FDI2 using identical haemoglobin- and weight-based dosing regimens at baseline and at Day 35.3
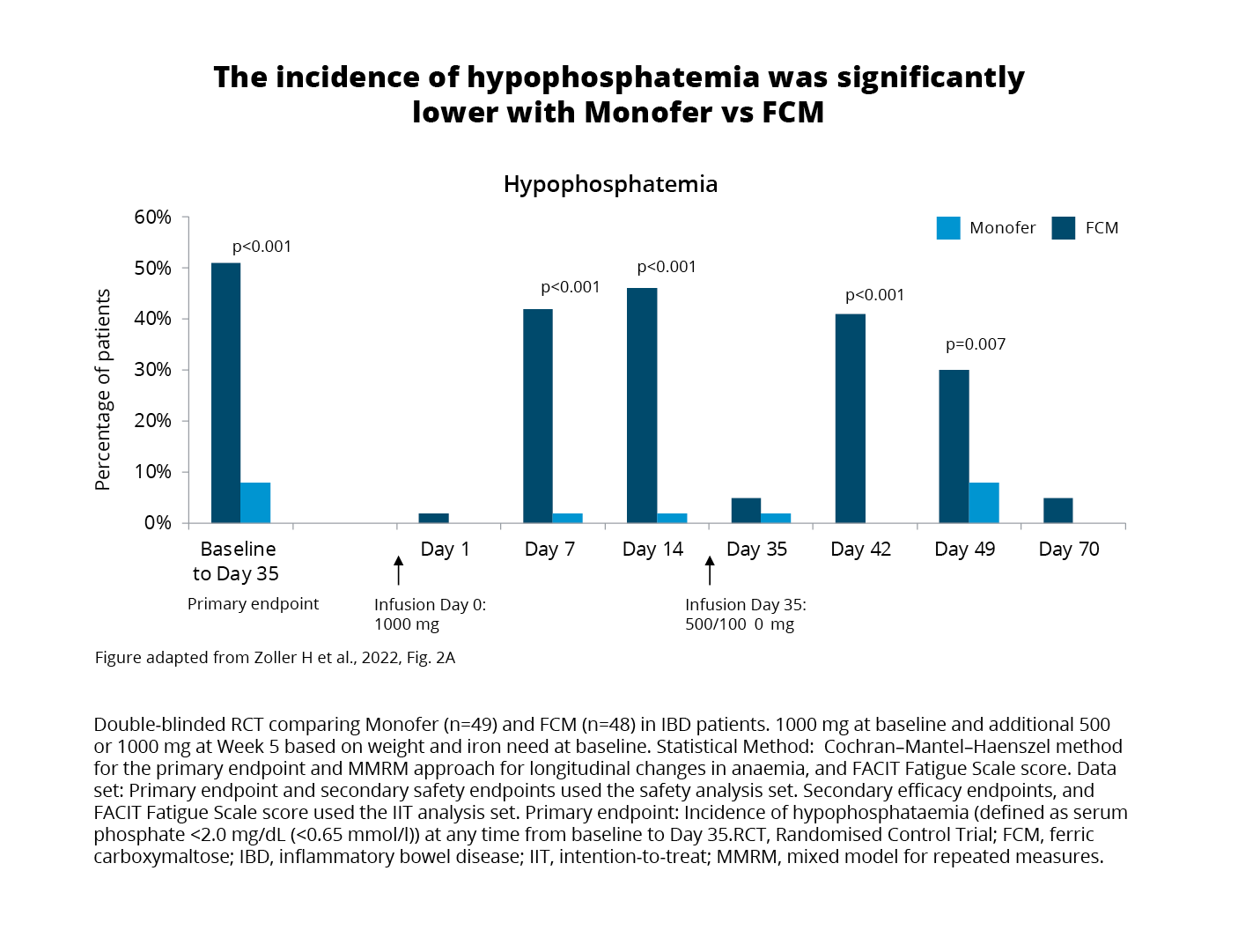
Exploring the primary endpoint using the safety analysis set (all patients who received at least one dose of the study drug) revealed comparable effects on haemoglobin (Hb) and iron stores after receiving either FCM1 or FDI2.3 On the contrary, hypophosphatemia (serum phosphate <2.0 mg/ dL (<0.65 mmol/l)) occurred in:3
(Figure 1)
When compared with FDI2, FCM1 treatment was associated with more pronounced changes in biomarkers showing increases in intact FGF23 and renal phosphate excretion, and PTH, and decreases in 1,25-dihydroxyvitamin D all related to an increased risk of hypophosphatemia.3
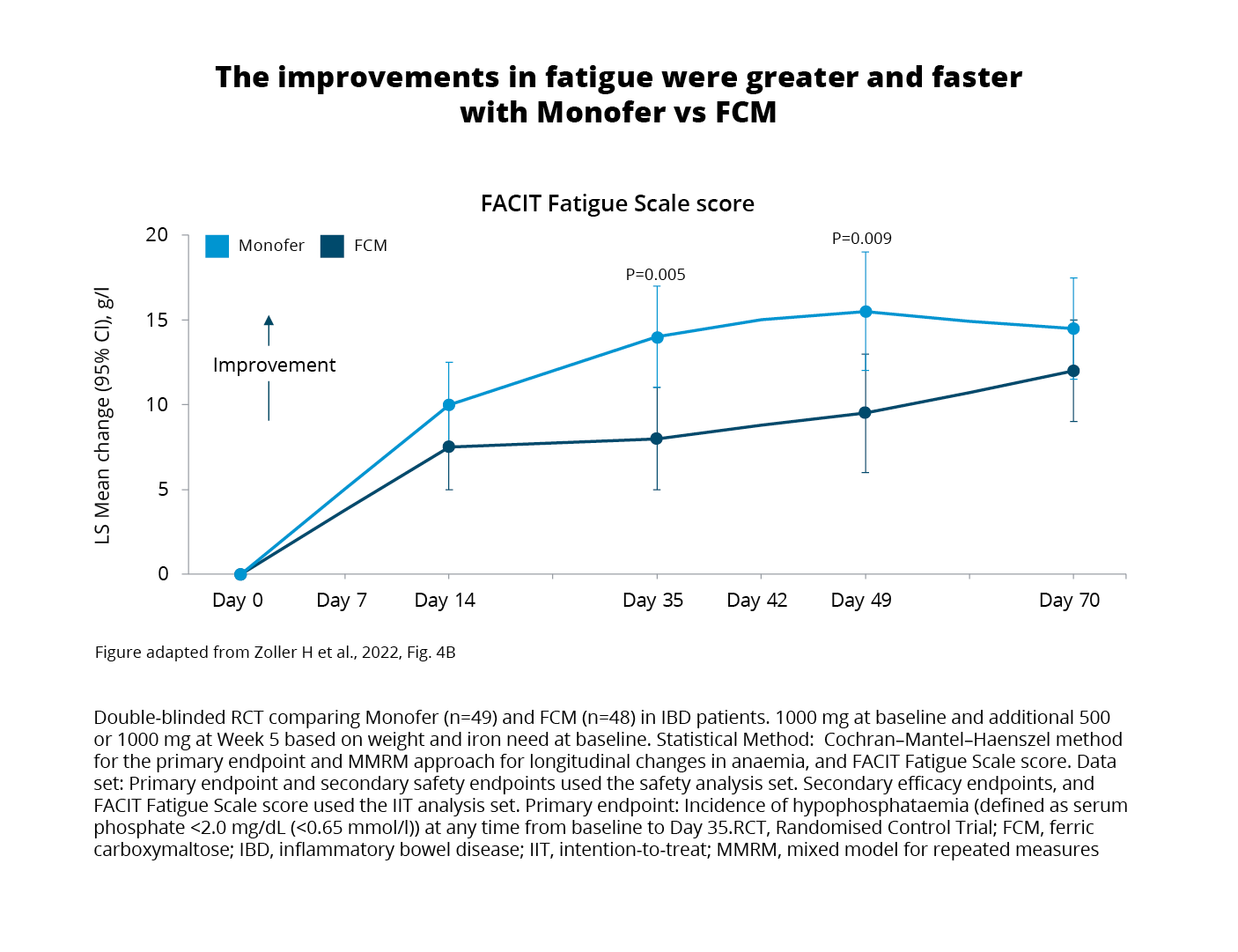
Analysis of patient-reported fatigue scores using the Functional Assessment of Chronic Illness Therapy (FACIT) Fatigue Scale was carried out within the intention-to-treat (ITT) population.3 The analysis aimed to explore the hypothesis of a potential relationship between phosphate level and fatigue.3 Results showed improved scores in both treatment groups. However, it is noteworthy that:
Despite recovering from hypophosphataemia following their initial treatment, patients treated with FCM1 still experienced prolonged biochemical changes linked to elevated FGF expression.3 Specifically, bone-specific ALP remained elevated on Day 70 and beyond implying a sustained impact on bone turnover.3
Based on the study design the results from the PHOSPHARE-IBD trial indicate that differences in hypophosphataemia are not a class or dose effect of IV iron or due to dosing, schedule, or patient factors but solely an adverse effect of FCM1 driven by increases in FGF23.3
Furthermore, the discovery of significant differences in patient-reported fatigue and the link to hypophosphataemia severity offers valuable insights that advance the field.3 This information could assist in guiding future treatments by informing clinicians and patients about the short-term effects of hypophosphataemia on quality of life and the risk of long-term effects on mineral and bone metabolism increasing the risk of osteomalacia and fractures.3
Disclaimer
Despite the dramatically different effects on plasma phosphate homeostasis, FDI2 and FCM1 treatment have both been consistently shown to improve quality of life in patients with iron deficiency. The present trial confirms that both formulations effectively corrected IDA, as evidenced by almost identical Hb responses leading to a rise of 20–30 g/L in both groups as well as correction of serum iron parameters.3
Note: Vennligst se den fullstendige preperatomtalen før produktet forskrives.
Legemiddelform: Infusjons-/injeksjonsvæske, jern(III)derisomaltose, oppløsning 100 mg/ml. Tilgjengelig som hetteglass: 5 x 1 ml, 5 x 5 ml og 2 x 10 ml. Indikasjoner: Behandling av jernmangel ved følgende indikasjoner: Ved klinisk behov for hurtig tilførsel av jern. Når orale jernpreparater ikke kan benyttes pga. manglende effekt eller ikke kan brukes av andre årsaker. Diagnosen må baseres på laboratorieprøver. Dosering: Doseringen gjøres trinnvis: [1] bestemmelse av det individuelle jernbehovet, og [2] utregning og administrasjon av jerndosen(e). Trinnene kan gjentas etter [3] vurdering av jernoppfylling etter jerntilskudd. Jernbehovet er uttrykt i mg elementært jern. Jernbehovet kan bestemmes enten ved en forenklet tabell basert på Hb-verdi og kroppsvekt eller ved Ganzoni-formelen (se preparatomtalen). For å vurdere effekten av Monofer skal Hb-nivået revurderes tidligst 4 uker etter siste administrering. Ved ytterligere behov for jern, må dette utregnes på nytt. Administrasjon: Preparatet gis som i.v. bolusinjeksjon, som i.v. infusjon eller som en injeksjon direkte i veneslangen ved hemodialyse. I.v. bolusinjeksjon: Doser opptil 500 mg pr. injeksjon opptil 3 ganger ukentlig og med injeksjonshastighet på opptil 250 mg jern/minutt kan tilføres. Dosen kan gis ufortynnet eller fortynnes. I.v. infusjon: Hele jerndosen, opptil 20 mg jern/kg kroppsvekt, kan gis som én engangsinfusjon eller som ukentlige infusjoner til det kumulative jernbehovet er gitt. Dersom jernbehovet overstiger 20 mg jern/kg kroppsvekt, må dosen deles opp i 2 administreringer med minst 1 ukes mellomrom. Det anbefales, når det er mulig, å gi 20 mg jern/kg i 1. administrering. Avhengig av klinisk skjønn kan 2. administrering avvente oppfølgende laboratorieprøver. Skal infunderes ufortynnet eller fortynnet, med steril 9mg/ml natriumkloridoppløsning, i maksimalt 500 ml. Kontraindikasjoner: Overfølsomhet for innholdsstoffene eller kjent overfølsomhet for andre parenterale jernpreparater. Anemi uten at det foreligger jernmangel (f.eks. hemolytisk anemi). For høyt jernnivå eller forstyrrelser i kroppens utnyttelse av jern (f.eks. hemokromatose, hemosiderose). Dekompensert leversykdom. Forsiktighetsregler: Parenteralt administrerte jernpreparater kan gi overfølsomhetsreaksjoner, inkl. alvorlige og potensielt dødelige anafylaktiske/anafylaktoide reaksjoner. Overfølsomhetsreaksjoner er også sett etter tidligere bivirkningsfrie doser av parenterale jernkomplekser. Overfølsomhetsreaksjoner som har utviklet seg til Kounis syndrom er sett. Risikoen er økt ved kjente allergier. Pasienten bør observeres for bivirkninger i minst 30 minutter etter hver administrasjon. Behandlingen må stoppes umiddelbart ved overfølsomhetsreaksjoner eller tegn på intoleranse under administrering. Parenteralt jern bør brukes med forsiktighet til pasienter med akutt eller kronisk infeksjon. Bør ikke gis til pasienter med aktiv bakteriemi. Overfølsomhets-reaksjoner kan oppstå, hvis IV injeksjonen gis for hurtig. Bivirkninger: Akutte, alvorlige overfølsomhetsreaksjoner kan forekomme ved administrering av parenterale jernpreparater. De opptrer vanligvis i løpet av de første minuttene etter infusjonsoppstart og er karakterisert ved plutselig innsettende pustebesvær og/eller sirkulatorisk kollaps. Vanlige: Kvalme, utslett, reaksjoner på injeksjonsstedet. Vennligst se den komplette preperatomtale for opplysninger om alle bivirkninger. Graviditet: Det er begrenset data på bruk av Monofer hos gravide kvinner, dette fra en studie med 100 behandlede gravide kvinner. En grundig nytte/risiko-vurdering er påkrevd før bruk under graviditet. Behandling med Monofer bør begrenses til andre og tredje trimester.
Ytterligere sikkerhetsinformasjon: Bør ikke brukes til barn og ungdom <18 år. Kan gi overfølsomhetsreaksjoner, inkl. alvorlige og potensielt dødelige anafylaktiske/anafylaktoide reaksjoner. De opptrer vanligvis i løpet av de første minuttene etter infusjonsoppstart og er karakterisert ved plutselig innsettende pustebesvær og/eller sirkulasjonssvikt. Overfølsomhetsreaksjoner som har utviklet seg til Kounis syndrom er sett. Risikoen er økt ved kjente allergier og ved autoimmune eller inflammatoriske tilstander. Behandlingen må stoppes umiddelbart ved overfølsomhetsreaksjoner eller tegn på intoleranse under administrering.
Pakninger og priser (per 09/22): ATC: B03AC. Hetteglass: 5 x 1 ml kr 1532,40. 5 x 5 ml kr 7203,10. 2 x 10 ml kr 5770,60.
Reseptgruppe C
Basert på SPC godkjent 11.08.2022.
For ytterligere informasjon om Monofer®, se SPC. www.felleskatalogen.no
Innehaver av markedsføringstilatelsen: Pharmacosmos A/S, Rørvangvej 30, DK- 4300 Holbæk, Danmark www.pharmacosmos.com.
07-SCAN-11-09-2023.v1
PHOSPHARE-IBD study presentation
Gastro advertorial Norway
Cardio advertorial Norway
Gastroenterology article 1



